Overview
The research in structural biology has accelerated to a large extent with the advent of multiple sequencing projects. The availability of huge amount of sequence data has increased the need for the implementation of different structural biology techniques. Biomolecular simulations involve in-silico techniques that examine the structural data obtained from experimental techniques. The group has gained experience in advanced simulation techniques like Replica Exchange Molecular Dynamics (REMD), Coarse Grained Molecular Dynamics (CGMD) and Classical MD song with advanced analytics techniques. The group works in the diverse areas and have multiple publications in international peer journals.
Protein folding studies
Protein folding can be defined as the understanding of how a newly synthesized polypeptide is able to fold to its native structure passing through various phases. The most optimum path through these phases is what defines the protein folding pathway. Emulating this activity in-silico, requires a large amount of computational power and time, even for the fastest folding protein, and even today in many ways this pathway remains a mystery. Advanced techniques such as REMD, Umbrella sampling techniques and cascade MD have employed on different protein. Along with this team is also trying to understand how misfolding of various protein leads to neurodegenerative diseases.
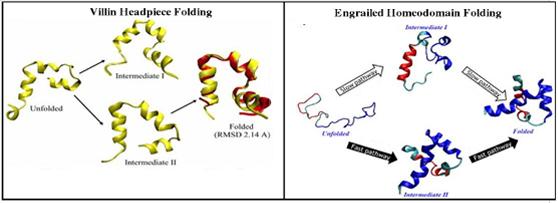
Figure : Protein folding pathway study for Villin and Engrailed homoedomain protein
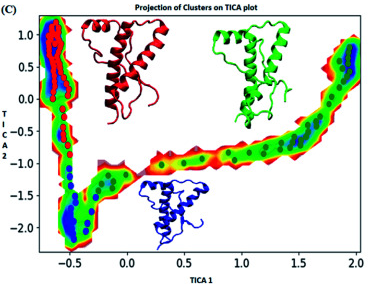
Figure : Markov state model analysis for E219K variant of prion protein responsible for neurodegenerative disease. Free energy landscape along first two time independent component showing three major states (red, blue and green) along with their representative structure.
RNA Therapeutics: Next-Generation Drug Discovery Technology
Antisense Oligonucleotides Simulations
The antisense technology has been explored and implemented for regulation of gene expression in animals, and identified as a potential tool for a novel therapeutic platform. The antisense molecules obstruct the gene expression and inhibit the synthesis of undesired proteins. The existing drugs directly interact with the disease-causing proteins throughout the body, as seen in traditional discovery, whereas the antisense based drugs inhibit the production of such proteins by directly targeting the mRNA involved. Antisense drugs are chemically modified oligonucleotides (Antisense Oligonucleotides: ASOs) that are complementary to target mRNAs and bind by Watson-Crick base pairing forming ASO-mRNA hetero-duplexes. mRNA bound by an ASO activates the RNase H enzyme which further cleaves the RNA strand selectively from the ASO-mRNA hetero-duplexes. The RNase H is specifically binds to DNA-RNA hybrid duplex and cleaves the mRNA chain. The mechanism of cleavage is identified in cellular functions and is being tried to explore through different structural studies. Understanding the mechanism of antisense technology and various antisense modifications through different structural studies is very helpful for designing novel antisense oligomers against various diseases. The computational and molecular dynamics simulations studies of antisense molecules are being carried out.
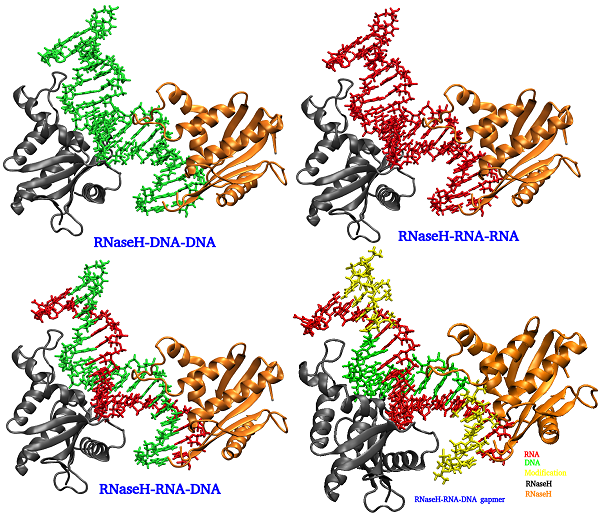
Figure: Duplexes bound to RNase H
RNAi (siRNA) Simulations
RNA interference (RNAi) technology is a promising approach to inhibit viral replication through a highly conserved, very efficient process of post-transcriptional gene silencing that is triggered by short double-stranded RNA, such as small interfering RNA (siRNA). One of the known mechanisms is observed in the bacterial defense system, where a foreign viral RNA (vRNA) enters into the cell, the bacterial siRNA is synthesized and the machinery to act on the viral RNA is built using the RNA-induced silencing complex (RISC). RNAi leads to the degradation of mRNA by binding to the target RNA in a sequence-specific manner and ultimately inhibits the expression of the specific genes. Several RNAi-based therapies have been investigated for the treatment of viral infections. Research studies demonstrated an effective reduction of target viral gene expression and viral replication in various cells and animal disease models like human immunodeficiency virus (HIV), influenza virus, coxsackievirus B3, adenovirus, hepatitis B virus, hepatitis C virus to name a few. RNAi is one of the efficient RNA based strategies to target viral diseases. Identification of siRNA target in viral genome, designing of siRNAs and understanding the mechanism help in discovering the novel siRNA based drugs for the viral diseases. The computational and structural studies are being carried out.
Structural studies of tRNA3Lys, an human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) primer
The human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) utilizes human tRNA3Lys as a primer for the formation of the reverse transcription initiation complex with the help of the nucleocapsid protein. It has been observed that either tRNA3Lys alone or HIV-1 RT or reverse transcription initiation complex were targeted by small molecules for inhibiting HIV-1. The tRNA3Lys was targeted with the RNA specific small molecules to inhibit the reverse transcription. The structural studies like molecular docking and molecular dynamics simulations were carried out. Peptide Nucleic Acid (PNA), one of the antisense molecules proves to be of great significance in HIV therapeutics. It has been observed that PNA oligomer targeted to the primer binding site (PBS) of viral RNA destabilize the crucial interactions between viral RNA primer and tRNA template due to which the reverse transcription is inhibited. The computational molecular dynamics simulations were carried out to understand the stability provided by the modified nucleotides of tRNA as well as the destabilization taken place due to presence of PNA oligomer.
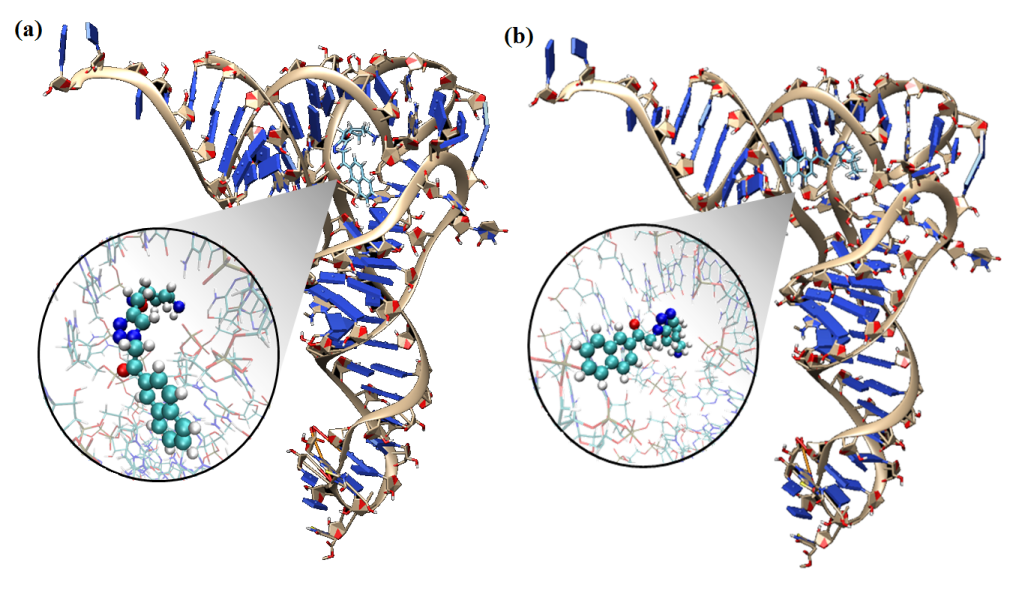
Figure: Binding poses of different ligands to tRNA3Lys
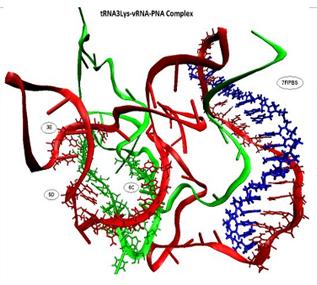
Figure: The complex structure of tRNA3Lys, viral RNA and PNA oligomer
Cancer Protein Studies
Cancer is a condition in which the cell undergoes uncontrolled growth and forms tumor. The proteins that perform their function normally undergo mutations to lose their functionality finally leading to cancerous conditions. The various cancer-related proteins that are being studied worldwide include cyclin-dependent kinases, p53, epidermal growth factor, RAS, pro-protein convertases and many more.
Tumor suppressor p53
The tumor suppressor p53 is one of the crucial proteins known to be mutated in 50 % of cancers. An alteration in p53 regulation or mutational defect in the protein functioning are known to occur in cancerous conditions. The conformations adopted in the lower temperatures would help in understanding the interactions crucial in maintaining the DNA binding. Through simulation studies of the DNA-free and DNA-bound form of p53 at different temperature, the temperature sensitive nature of protein has been studied . Enhanced analysis like Markov State Modeling has been used to identify conformational signatures. The findings obtained through this work would be beneficial in designing therapeutics for rescuing p53 in many different types of cancers.
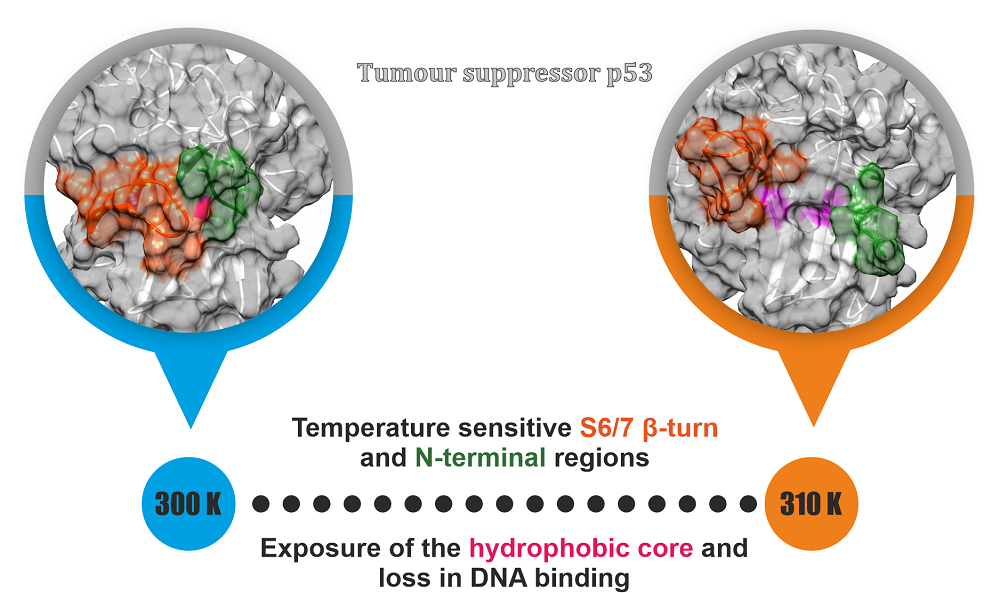
Figure: Temperature sensitive nature regions of p53 that can be used for designing therapeutics
Ras Oncogene
Ras pathway is one of the most crucial pathways of humans, the malfunctioning of which leads to oncogenic state and ultimately cancer. KRAS protein is known to be frequently mutated in various cancers. The most common mutations being at position 12, 13 and 61. The positions 12 and 13 form part of the phosphate binding region (P-loop) of KRAS. Owing to mutation, the protein remains in continuous active state and affects the normal cellular process. Understanding structural changes owing to mutations in GDP-bound (inactive state) and GTP-bound (active state) may help in the design of better therapeutics. Extensive molecular simulation and advanced analytics have been used for understand the structural flexibility due to mutations.
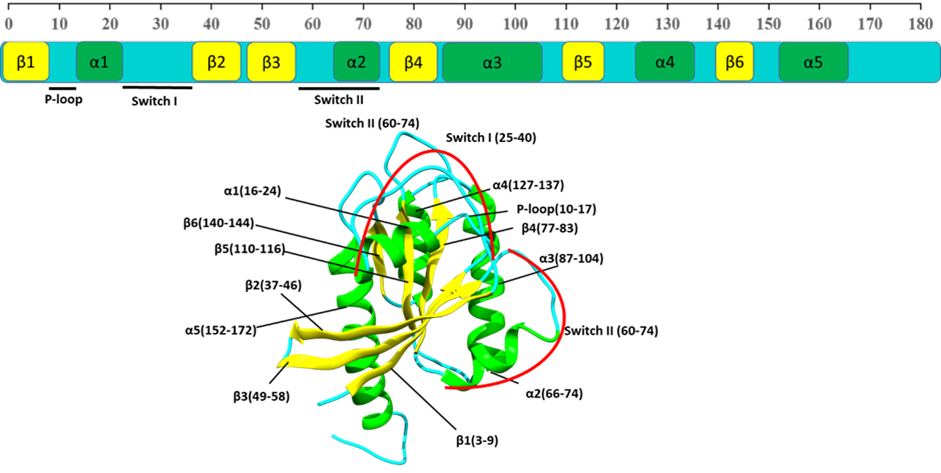
Figure: Structure of wild-type (WT) KRAS depicting different secondary structure elements and their sequence.
Membrane Protein Studies
Membrane protein, in particular GPCRs, constitutes a major portion of human protein, being directly involved in several signaling pathways and thus are paramount pharmaceutical interest. The process of GPCR dimer or oligomer formation, and its effect on receptor function, is not currently well understood. Coarse grained molecular dynamics (CGMD) approach has been adopted for studying the self-assembly process of the human amine GPCR protein β2-adrenergic receptor, for which several experimental evidences of oligomerization process and its effects on its function are available.
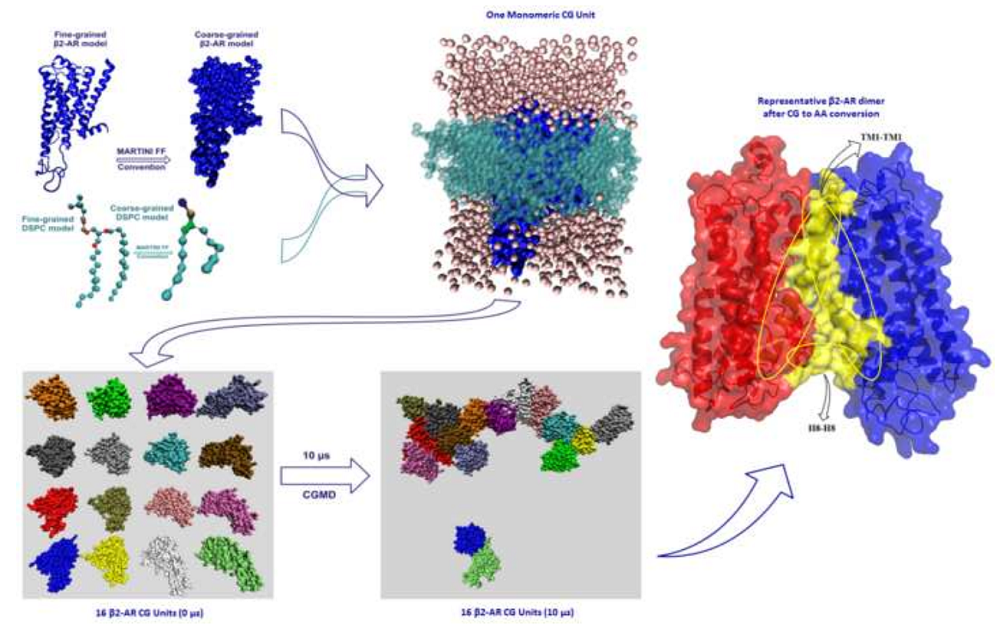
Figure : Multiscale modelling to understand the self-assembly mechanism of human β2-adrenergic receptor in lipid bilayer Author
β2AR can exist as a monomer, dimer and various oligomeric states; various ligands are able to activate this receptor and exert their physiological responses by triggering various G-proteins. There are evidences for dimer level constitutive activation of β2AR, so understanding the activation mechanism of β2AR at dimer level will give more insights about structural reaarangements of TM helices and gives us clues for novel drug designing aspects and thus activation mechanism at dimer level is being explored.
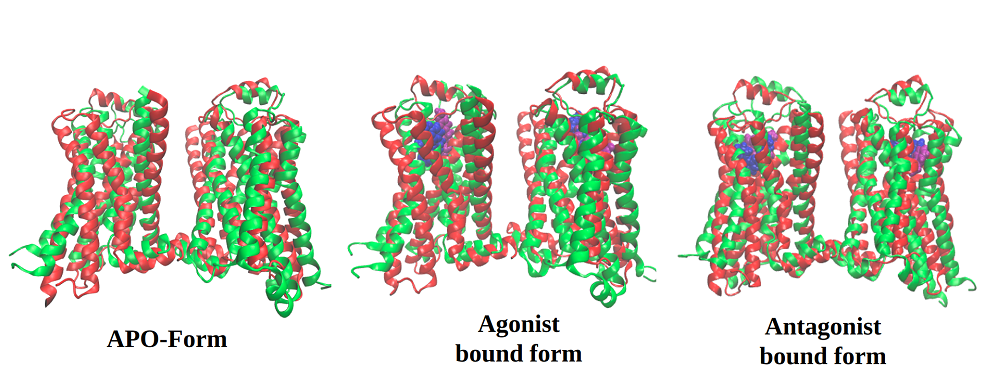
Figure: Understanding activation mechanism of β2AR dimer by comparative molecular dynamic simulations using apo, agonist and antagonist bound states